A Monte
Carlo módszer alapjai
A
számítógépes szimulációs módszerek az anyagi rendszer mikroszkopikus
tulajdonságainak, azaz a molekulák vagy atomok közötti
kölcsönhatásoknak az
ismeretében a sokrészecskés rendszer mikroállapotait közvetlenül
modellezik és
a fázistérből ily módon mintát véve a keresett tulajdonságokat sokaság-
vagy
időátlagként számítják. Az intermolekuláris potenciálokon kívül szükség
van még
néhány termodinamikai állapotjelző rögzítésére a használt sokaságtól
függően.
Két alapvető szimulációs módszer létezik, az egyik a molekuláris
dinamikai
(MD), a másik a Monte Carlo (MC) módszer. A MD szimulációk során a
rendszer
fázistérbeli trajektóriáját a klasszikus newtoni mozgásegyenletekkel
határozzák
meg. A trajektória mentén számított fizikai mennyiségek átlaga
időátlagnak
tekinthető MD szimulációk során.
A
disszertációban a Monte Carlo módszert alkalmaztuk, ezért ezt
részletesebben
ismertetjük. A Monte Carlo szimulációk során véletlenszerűen veszünk
mintát a
konfigurációs tér pontjai közül, így különböző mikroállapotú rendszerek
sokaságát
állítjuk elő. A módszer nem alkalmas nemegyensúlyi, időben változó
rendszerek
vizsgálatára, csak az egyensúlyban levő rendszerek sztatikus jellemzői
határozhatóak meg. A részecskék „mozgása” indeterminisztikus,
valószínűségi
törvénynek engedelmeskedik.
A
módszer alapjait a kanonikus sokaságon ismertetjük. Tekintsünk egy V térfogatú, kocka alakú szimulációs
cellát, amely N részecskét tartalmaz.
Esetenként több százezres nagyságrendű részecskeszámmal is végeznek
szimulációkat,
de a minta még így sem tekinthető makroszkopikusnak. Az oka a
következő: a
szimulációs doboz határfelületén nagyon sok részecske helyezkedik el,
így a
határfelületi jelenségek szerepe jelentős. A periodikus
határfeltétel alkalmazásával kiküszöbölhetőek a
határfelületi jelenségekből származó hibák, mivel a középpontinak
tekintett
cella körül ebben az esetben végtelen számú ugyanolyan cella
helyezkedik el.
Ezekben a cellákban a szellemrészecskék ugyanúgy mozognak, mint a
központi
cella részecskéi. Ez azt jelenti, hogy ha egy részecske kilép a
kockából egy
adott irányban, a szomszéd cellából belép a megfelelő „szellemrészecske” az ellentétes irányból.
Valamely
konfigurációs fizikai mennyiség értéke a
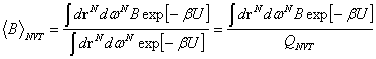
egyenlet
szerint adott. A nevezőben a ![]() kanonikus
konfigurációs integrál
található. Az
integrálok megbecsülhetőek úgy, hogy a konfigurációs tér
elegendően sok
kanonikus
konfigurációs integrál
található. Az
integrálok megbecsülhetőek úgy, hogy a konfigurációs tér
elegendően sok ![]() pontjában kiszámítjuk
pontjában kiszámítjuk ![]() és
és ![]() értékét, így az
integrált összegzéssel helyettesítjük:
értékét, így az
integrált összegzéssel helyettesítjük:
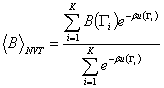
ahol K a
mintapontok száma. A MC szimulációk
során a teljes konfigurációs térből kell egyenletesen mintát venni majd
azt a
Boltzmann faktorral súlyozva figyelembe venni. Ez az eljárás még mindig
meghaladja a számítógépek teljesítőképességét.
A
számítási idő jelentősen csökkenthető, ha a mintát nem egyenletes
eloszlás
szerint vesszük, azaz ha egy adott ![]() pont valamely
pont valamely ![]() eloszlásnak megfelelő
valószínűséggel kerül kiválasztásra. Az ilyen mintavétel során csak
azokra a
konfigurációs pontokra koncentrálunk, amelyek jelentős járulékot adnak
az
állapotösszeghez. A fenti átlagban a súlyozást kompenzálni kell, így:
eloszlásnak megfelelő
valószínűséggel kerül kiválasztásra. Az ilyen mintavétel során csak
azokra a
konfigurációs pontokra koncentrálunk, amelyek jelentős járulékot adnak
az
állapotösszeghez. A fenti átlagban a súlyozást kompenzálni kell, így:
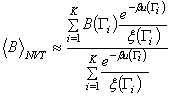
Ha a mintavételnél
alkalmazott eloszlás a Boltzmann-eloszlás ![]() ,
akkor Boltzmann-mintavételről beszélünk, vagyis az
átlagolásnál azonos súllyal vesszük figyelembe a számolt értékeket:
,
akkor Boltzmann-mintavételről beszélünk, vagyis az
átlagolásnál azonos súllyal vesszük figyelembe a számolt értékeket:
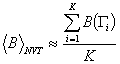 .
.
A Metropolis
féle mintavételezés lényege a
következő. A mintapontokat Markov lánc tagjainak tekinti, ahol annak a
valószínűsége,
hogy ![]() bekerül a mintába csak
a lánc előző tagjától függ. Ha
bekerül a mintába csak
a lánc előző tagjától függ. Ha ![]() és
és ![]() lehetséges állapotai a
rendszernek és az ehhez tartozó Boltzmann faktorok
lehetséges állapotai a
rendszernek és az ehhez tartozó Boltzmann faktorok ![]() és
és ![]() , akkor az i
állapotból j-be való átmenet valószínűsége (
, akkor az i
állapotból j-be való átmenet valószínűsége (![]() )
egy sztochasztikus mátrixot definiál, amelyre a következő
feltételek teljesülnek:
)
egy sztochasztikus mátrixot definiál, amelyre a következő
feltételek teljesülnek:
![]() és
és
![]() minden
i-re.
minden
i-re.
Egy
adott kezdeti állapotból kiindulva a Markov folyamat segítségével
állítjuk elő
az egymás után következő állapotok sorozatát, amelyet a fenti átmeneti
mátrix irányít.
A mátrix sajátvektora a Boltzmann-eloszlás által meghatározott ![]() határeloszlás,
amelynek sajátértéke egységnyi. Ehhez az ismert határeloszláshoz olyan
átmeneti
mátrixot kell találni, amely kielégíti a fenti feltételeket, valamint a
mátrixelemek függetlenek az állapotösszegtől. Az említett feltételeket
pl. a
következő konstrukció elégíti ki:
határeloszlás,
amelynek sajátértéke egységnyi. Ehhez az ismert határeloszláshoz olyan
átmeneti
mátrixot kell találni, amely kielégíti a fenti feltételeket, valamint a
mátrixelemek függetlenek az állapotösszegtől. Az említett feltételeket
pl. a
következő konstrukció elégíti ki:
![]()
Ezt nevezik
a mikroszkopikus reverzibilitás feltételének is, és lényege az, hogy
egyensúlyban a szimulációban az i
állapotból a j-be jutás valószínűsége
ugyanakkora, mint a j állapotból az i-be
jutás valószínűsége. Az átmeneti
valószínűség két tag szorzataként áll elő:
![]()
ahol aij annak
valószínűsége, hogy a szimuláció során az i
állapot után a j állapotot sorsoljuk,
míg Pij annak a
valószínűsége, hogy az i állapotból a
j állapotba való mozgatást
elfogadjuk. Ha az aij mátrix
szimmetrikus, mint a mi szimulációink esetében (nem feltétlenül kell
szimmetrikusnak lennie), akkor írhatjuk, hogy:
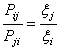
Metropolis
és munkatársai a következő megoldást adták a problémára [57-60]:

Az ![]() mátrixra a következő
algoritmust alkalmazhatjuk. Megpróbálunk a kocka belsejében egy
véletlenszerűen
kiválasztott részecskét véletlenszerűen elmozgatni. A részecske új
helyét egy
egyenletes eloszlást produkáló véletlenszám-generáló rutin segítségével
sorsoljuk a következő módon:
mátrixra a következő
algoritmust alkalmazhatjuk. Megpróbálunk a kocka belsejében egy
véletlenszerűen
kiválasztott részecskét véletlenszerűen elmozgatni. A részecske új
helyét egy
egyenletes eloszlást produkáló véletlenszám-generáló rutin segítségével
sorsoljuk a következő módon:
![]()
![]()
![]()
ahol ![]() a (0,1) intervallumban
egyenletesen generált véletlenszámok. Ez azt jelenti, hogy a részecskét
egy a
régi hely körüli 2Drmax
élhosszúságú kockán belül egy véletlenszerűen kiválasztott pontba
áthelyezzük.
Ha Drmax
kicsi, akkor a részecske új helye a régihez közel van. Ez különösen
hasznos
folyadékokban, valamint a polarizálható fluidumok esetében, ahol az
indukált
dipólusmomentumok újraszámolását végző iteratív rutin gyorsabban
konvergál, ha
az indukált dipólusmomentumok átrendeződését generáló változás, azaz a
részecske elmozdulása kicsi. Ha a rendszer sűrűsége kicsi (gáz vagy híg
oldat),
a részecske új pozícióját sorsolhatjuk véletlenszerűen a teljes
szimulációs
cellában a régi pozíciótól teljesen függetlenül.
a (0,1) intervallumban
egyenletesen generált véletlenszámok. Ez azt jelenti, hogy a részecskét
egy a
régi hely körüli 2Drmax
élhosszúságú kockán belül egy véletlenszerűen kiválasztott pontba
áthelyezzük.
Ha Drmax
kicsi, akkor a részecske új helye a régihez közel van. Ez különösen
hasznos
folyadékokban, valamint a polarizálható fluidumok esetében, ahol az
indukált
dipólusmomentumok újraszámolását végző iteratív rutin gyorsabban
konvergál, ha
az indukált dipólusmomentumok átrendeződését generáló változás, azaz a
részecske elmozdulása kicsi. Ha a rendszer sűrűsége kicsi (gáz vagy híg
oldat),
a részecske új pozícióját sorsolhatjuk véletlenszerűen a teljes
szimulációs
cellában a régi pozíciótól teljesen függetlenül.
A
Boltzmann-eloszlást helyettesítve ![]() helyébe akkor fogadjuk
el az elmozdítást, ha az összenergia csökkent a folyamat során. Ha ez
nem áll
fenn, akkor az elmozdítás elfogadásának valószínűsége:
helyébe akkor fogadjuk
el az elmozdítást, ha az összenergia csökkent a folyamat során. Ha ez
nem áll
fenn, akkor az elmozdítás elfogadásának valószínűsége:
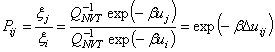
Látható, hogy az
algoritmus szükségtelenné teszi az
állapotösszeg kiszámítását. Ha az intermolekuláris potenciál nem
gömbszimmetrikus,
akkor a molekulák orientációját, azaz a polárszögeket is
véletlenszerűen meg
kell változtatni valamely ![]() határokon belül.
határokon belül.
Egy részecske kölcsönhatási energiájának számításakor azon L élhosszúságú kockában levő részecskéket kell figyelembe venni, amelynek a középpontjában az adott részecske helyezkedik el. A részecske energiáját szférikus levágás alkalmazásával kapjuk meg, vagyis az rc (ahol rc általában L/2-vel egyenlő) sugarú gömbön belül levő részecskékkel vett párkölcsönhatási energiákat összegezzük, míg a fennmaradó, gömbön kívül eső részecskék hatását hosszútávú korrekciókkal vesszük figyelembe. Ennek számítására a rövid hatótávolságú potenciálok (mint például a LJ potenciál) esetén pontos közelítő módszer áll rendelkezésünkre. Feltételezzük, hogy a párkorrelációs függvény egységnyi a központi részecskétől rc-től nagyobb távolságban, így az energia hosszútávú korrekciója (LRC, Long Range Correction) a következő módon számítható:
![]() .
.
Lennard-Jones potenciál esetén az integrálást elvégezve:
 .
.
Dipólus-dipólus kölcsönhatás esetén a potenciál hosszú hatótávolságú és irányfüggő. A hosszútávú korrekciók kezelésére többféle módszert választhatunk. A két legfontosabb a reakciótér [61] és az Ewald-Kornfeld összegzési módszerek [62].
A reakciótér módszer (amit a dolgozatban használunk) lényege a következő [61]. Az rc sugarú gömb középpontjában levő dipólus energiájának számításakor a gömbön kívül levő dipólusokat egy eRF dielektromos állandójú folytonos közeggé „mossuk össze”, és a központi dipólusnak ezekkel való kölcsönhatását, azaz a hosszú távú korrekciót a dipólus és a reakciótér kölcsönhatásaként közelítjük. A reakciótér a gömbben levő összes dipólus által a minta és az azt körülvevő dielektrikum határfelületén indukált polarizációs töltések által kifejtett erő. Erről részletesebben a 2.3 fejezetben volt szó, a reakciótérrel való kölcsönhatást a következő egyenlet definiálja:
![]() ,
,
ahol M
az ![]() sugarú mintában
(melynek középpontjában a
sugarú mintában
(melynek középpontjában a ![]() dipólus helyezkedik
el) levő összes dipólusmomentum.
dipólus helyezkedik
el) levő összes dipólusmomentum.
Ahogy
azt a
2.3 fejezetben kifejtettük, a határfeltételtől, azaz ![]() –től
függ a reakciótér, a dielektromos állandó és a
Kirkwood-faktor közti kapcsolat, külső tér alkalmazása esetén a
létrejövő
polarizáció is. Ugyanakkor függetlenek az alkalmazott határfeltételtől
az
állapotfüggvények és a dielektromos állandó is.
–től
függ a reakciótér, a dielektromos állandó és a
Kirkwood-faktor közti kapcsolat, külső tér alkalmazása esetén a
létrejövő
polarizáció is. Ugyanakkor függetlenek az alkalmazott határfeltételtől
az
állapotfüggvények és a dielektromos állandó is.
Az Ewald-Kornfeld szummázás [62] során kiszámítják a részecske kölcsönhatási energiáját az összes többi, szomszédos dobozban elhelyezkedő szellemrészecskével. Ez az összegzés is csak véges rendszerre végezhető azonban el és a (nagyobb) rendszert szintén dielektrikum veszi körül: ekkor fellép egy ún. felületi tag, de az ebből származó hiba az esetek többségében elhanyagolható.