
About Us
The macroscopic (measurable) properties of many-particle systems are determined by microscopic processes. Understanding these processes is possible by modelling the particles and their interactions on the molecular level and by studying these models with statistical mechanical methods. The research group use both analytic theories and computer simulations (molecular dynamics, Monte Carlo) for this purpose. The systems studied by the group include selective silicates (zeolites) usable for component separation processes, ion channels of the cell membrane, ion-selective and rectifying nanopores, and complex liquids (showing ordering and phase transitions). Novel simulation methods are being developed in order to study these systems. A 104-core computer cluster is available to run the time-consuming calculations. The members of the research group are recognized experts in the field of their research interest having diverse network of collaborators all over the world. They won several OTKA and other grants; they are keen on expanding the financial background for their research. They regularly present their results at international conferences and publish them in prestigious scientific journals.Recent Publications
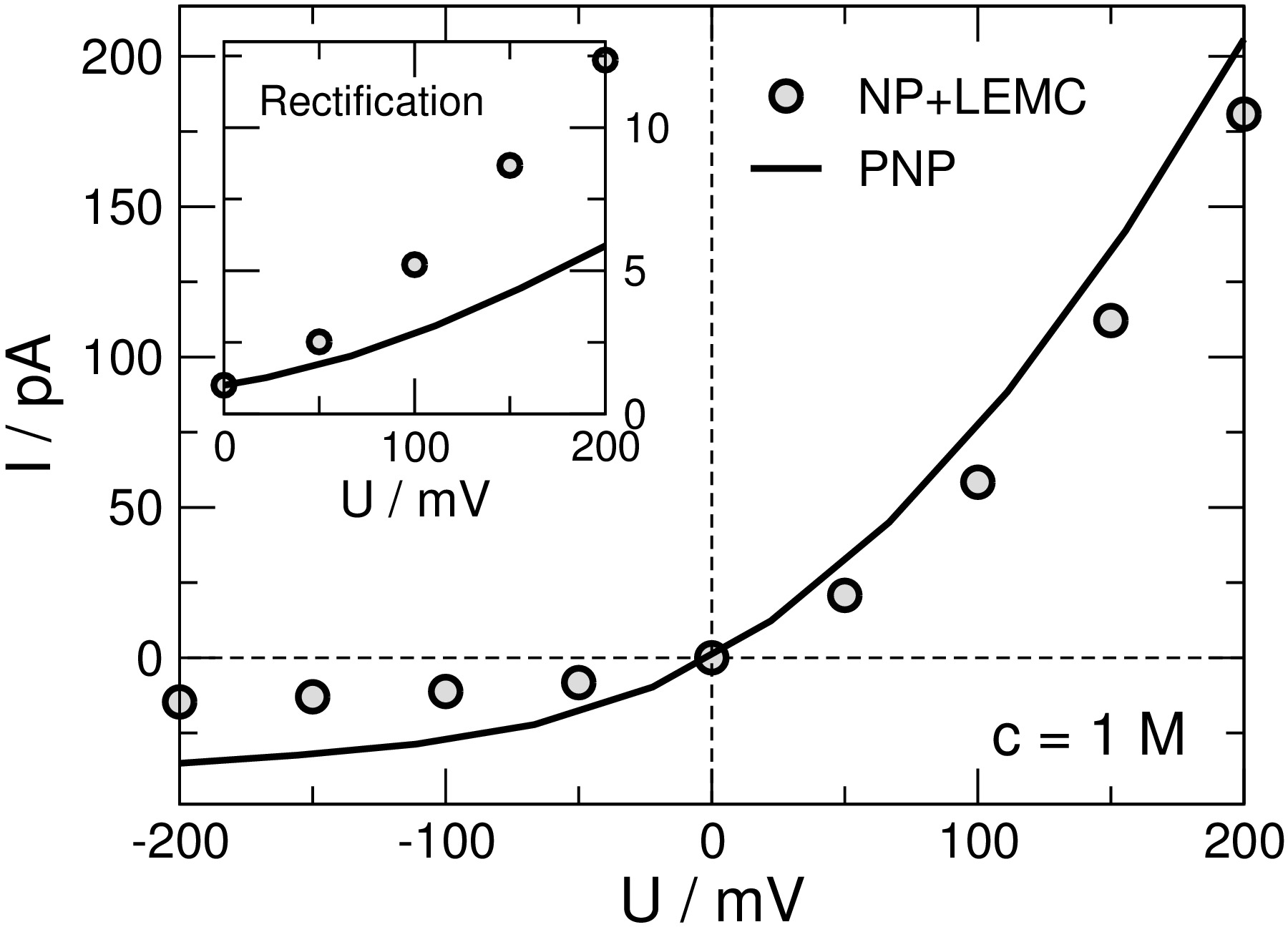
Comparing Poisson-Nernst-Planck to Monte Carlo
B Matejczyk, M Valiskó, M-T Wolfram, J-F Pietschmann, D Boda
The Journal of Chemical Physics 146 (12), 124125
2017
doi
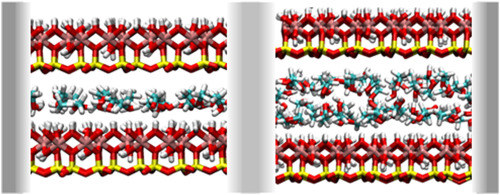
explicit-water versus implicit-water simulations
Z Ható, M Valiskó, T Kristóf, D Gillespie, D Boda
Physical Chemistry Chemical Physics [Accepted Manuscript]
2017
doi
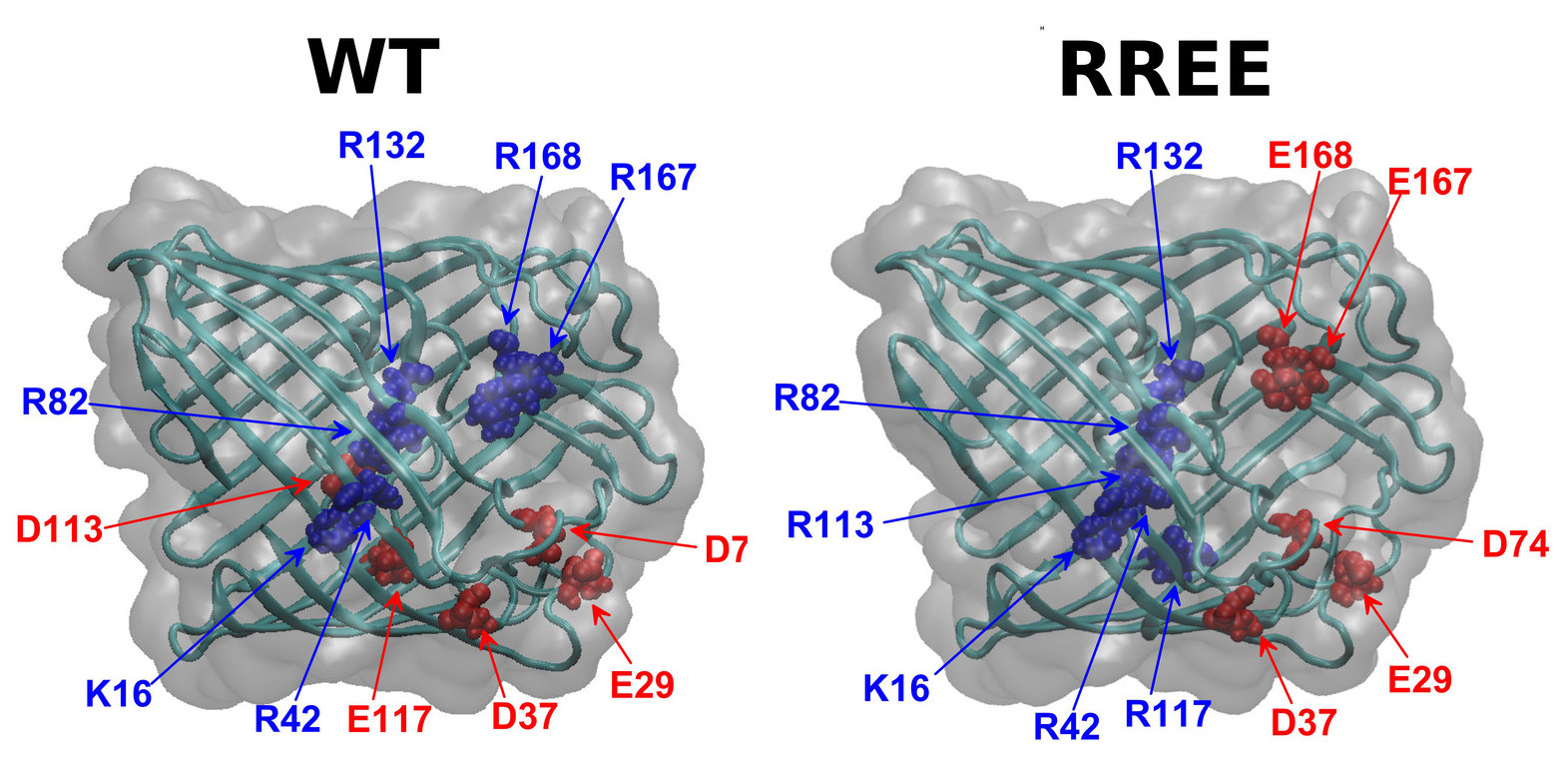
Detailed model versus reduced model
Z Ható, D Boda, D Gillespie, J Vrabec, G Rutkai, T Kristóf
Condensed Matter Physics 19 (1), 13802
2016
doi
A molecular simulation study
G Rutkai, Z Ható, T Kristóf
Fluid Phase Equilibria 409, 434-438
2015
doi
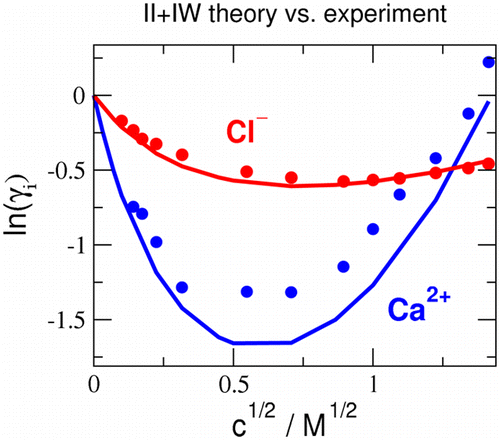
the basis of the balance of ion–ion and ion–water interactions
M Valiskó, D Boda
The Journal of Physical Chemistry B 119 (4), 1546-1557
2015
doi
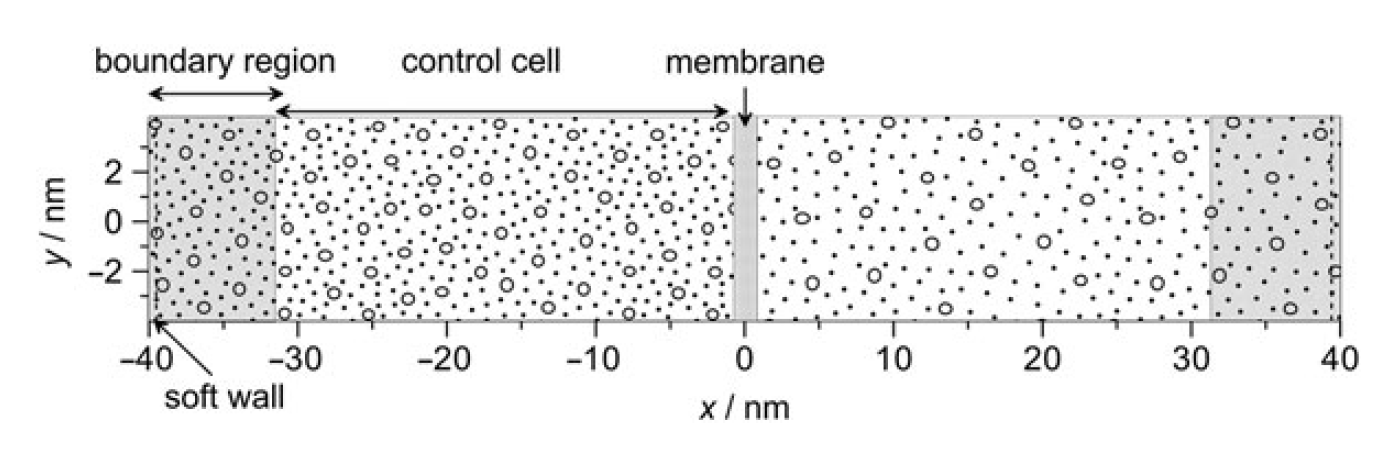
membranes: pressure-tuned, boundary-driven molecular dynamics
Z Ható, Á Kaviczki, T Kristóf
Molecular Simulation 42 (1), 71-80
2015
doi
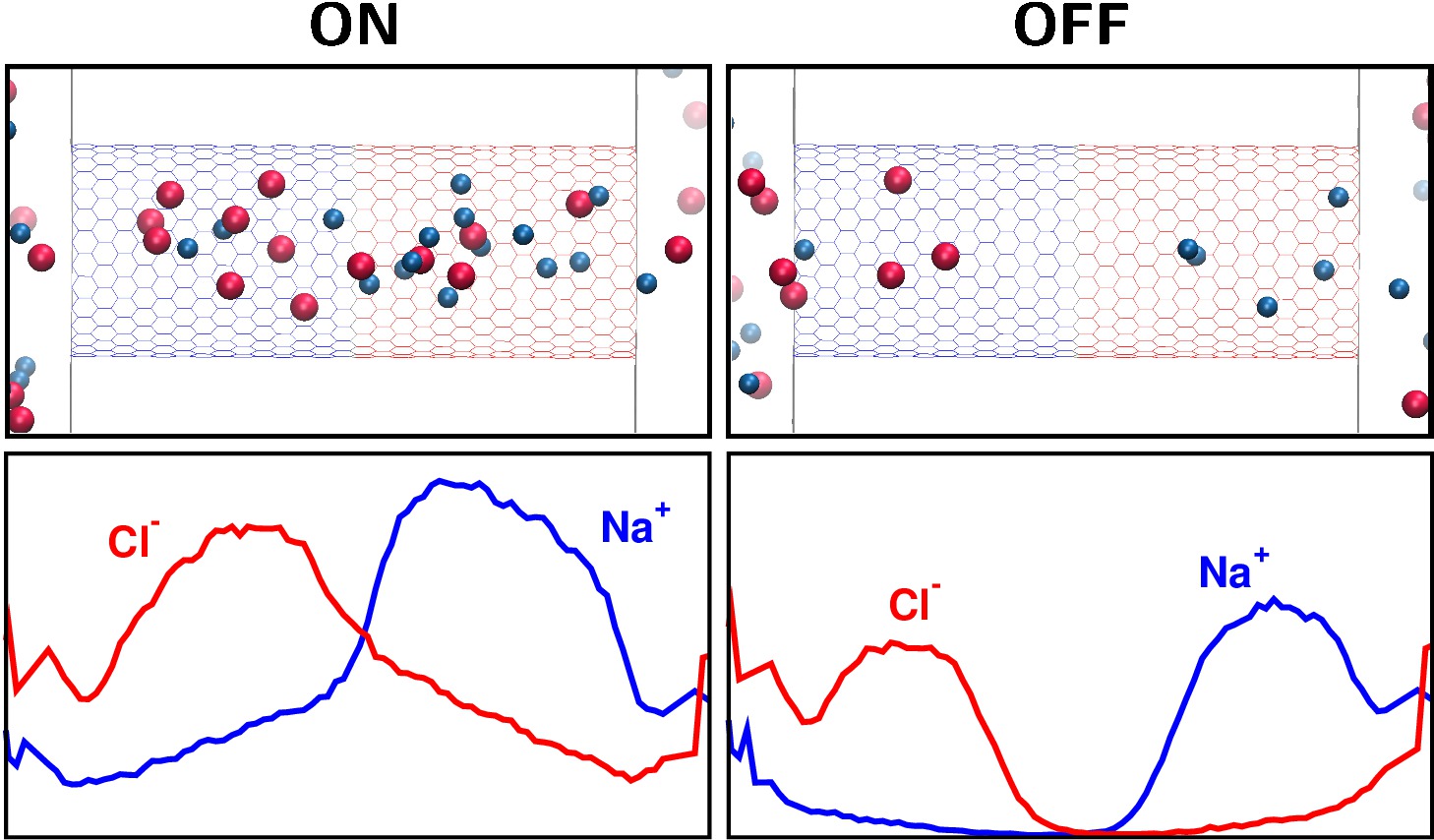
Rectifying bipolar nanopore
A molecular dynamics (MD) video clip showing ions (Na+ and Cl-) crossing through a bipolar nanopore in the ON (200 mV) and in the OFF (-200mV) state. The wall of the nanopore carries positive and negative charges. Depletion zone of a given ionic species is formed in the region, where the surface charge repels that ionic species (Na+ on the left and Cl- on the right). In the OFF state, the depletion zones are deeper, so the current is smaller. Therefore, the nanopore rectifies. In our recent work, we apply models of different resolutions for this system (all-atom models vs. reduced models) and use different techniques (MD, Nernst-Planck equation, Monte Carlo, Brownian Dynamics, Poisson-Nernst-Planck theory) to study this system in a multiscale modeling framework. Our results show that reduced models are useful in describing the nanopore's behavior as a device.



